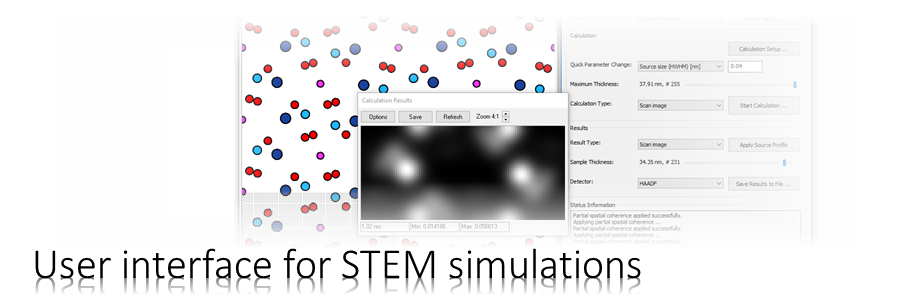




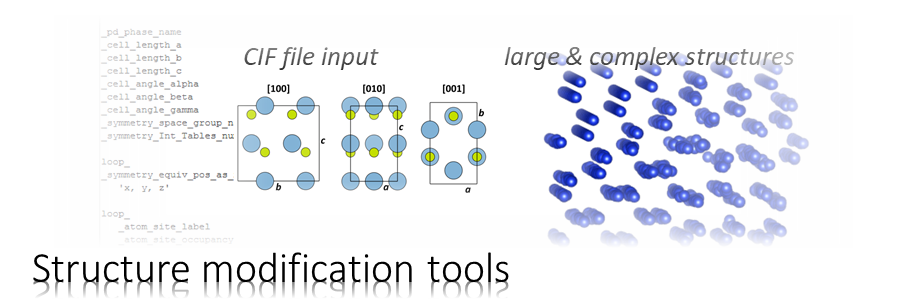
The program CELSLC is used to calculate object transmission functions aka. phase gratings from atomic structure models. Files describing the atomic structure and other parameters are specified as arguments upon calling. The output is oganized in so-called slice files, which are input for multislice calculations with the program MSA or the Dr. Probe GUI.
Calling the program CELSLC from a command-line shell, requires several command-line arguments providing important calculation parameters. The following table desscribes the command-line options and switches of CELSLC version 0.51 or higher.
| Option | Descriptions (Version 0.50+) |
| -cel <file name> | Specifies the input super-cell file containing the atomic structure data in CEL file format. Absolute or relative file names can be specified. Enclose the file name string by quotation marks if the file name contains space characters. Example: -cel 'cel\cell 1.cel' |
| -cif <file name> | Specifies the input super-cell file containing the atomic structure data in CIF file format. Absolute or relative file names can be specified. Enclose the file name string by quotation marks if the file name contains space characters. Example: -cif 'cel\crystal 2.cif' |
| -prj <u,v,w,u,v,w,a,b,c> | Re-orientation and re-sizing of the input structure model assuming a periodic crystal structure into an orthorhombic target cell. The orientation is given by the first 6 numbers, where the first tripple [uvw] defines the new projection direction in the input real-space lattice basis and the second triple [uvw] defines a perpendicular direction for the new y-axis of the projection. The size of the new orthorhombic super-cell is given by the last 3 numbers (a,b,c) defining the extension in nanometers along each of the 3 new axes x, y, and z, where z is the projection direction of the similation. The origin point of the structure is not changed. |
| -tla <fx,fy,fz> | Shift all atoms of the structure in the final orthorhombic super-cell by a given amount in fractional coordinates (fx,fy,fz). All atoms are shifted by the same amount and their final fractional coordinates are wrapped back periodically into the range [0,1). Example: -tla 0,0.125,0.0625 |
| -slc <file name> | Specifies the output slice file name prefix. Absolute or relative path names can be used. Enclose the file name string by quotation marks if the file name prefix or the disk path contains space characters. The slice file names will be suffixed by '_###.sli', where ### is a 3 digit number denoting the sequence of slices generated from the super-cell.Example: -slc 'slc\slices 1' |
| -nx <number> | Number of horizontal samples for the phase gratings, defining the minimum grid size of the multislice calculation. Avoid number with prime factors above 5 for keeping the calculations fast. Example: -nx 768 |
| -ny <number> | Number of vertical samples for the phase gratings, defining the minimum grid size of the multislice calculation. Avoid number with prime factors above 5 for keeping the calculations fast. Example: -ny 512 |
| -ht <number> | Accelerating voltage defining the kinetic energy of the incident electron beam in keV. Example: -ht 300.0 |
| -rev | (Optional) Switch reversing the order in which slices are generated from the input structure. When the option -rev is omitted, the slice sequence begins at the fractional z-coordinate z/c = 0 and stops at z/c = 1. When the option -rev is set, the slice sequence begins at z/c = 1 and stops at z/c = 0. When changing this option, the electron diffraction will not only be reversed along the z-coordinate, it will also change the rotational sense. |
| -fl | (Optional) Switch for applying random atomic displacements for frozen-lattice caluclations according to the specified thermal vibration parameter (Debye-Waller factor) B. This option cannot be used in combination with the options -dwf and -abs. |
| -nv <variant number> | (Optional) Specifies the number of frozen-lattice variants with random atom displacements generated per slice. Each slice file will contain several phase gratings of the same slice. Make sure to precalculate the required memory for stroring multiple variants. Do not exceed the physical size of the available RAM! Requires the option flag -fl.Example: -nv 25 |
| -nz <number> | (Optional) Equidistant slicing of the super-cell along the c-axis. Specify an explicit number of slices. Use -nz 0 to let CELSLC determine the number of equidistant slices automatically. Omitting the option -nz will lead to an automatic non-equidistant slicing.Example: -nz 4 |
| -ssc <number> | (Optional) Single slice calculation mode. Only the specified slice number will be calculated. |
| -dwf | (Optional) Switch for applying Debye-Waller factors which effectively dampen the atomic scattering potentials. Use this option for conventional HR-TEM, STEM bright-field, or STEM annular bright-field image simulations only. |
| -abs | (Optional) Switch for applying absorption potentials (imaginary part) according to Weickenmeier and Kohl [Acta Cryst. A47 (1991) p. 590-597]. This absorption calculation considers the loss of intensity in the elastic channel due to thermal diffuse scattering. The option -abs cannot be used in combination with the options -fl and -abf. |
| -abf <number> | (Optional) Switch and value for applying absorption potentials. The absorption potential is set as imaginary part of the scattering potential, which is a copy of the potential real part multiplied by a factor given by the specified floating point number. A typical value is 0.1. Example: -abf 0.085 |
| -pot | (Optional) Switch for exporting projected potential slices to individual files ( *.pot). Each slice variant will be saved to its own file. This may lead to output of a large number (nz*nv) of files. |
| -pps | (Optional) Switch writing projected potentials to slice files instead of phase gratings. This allows to apply absorption via -abf and uniform Debye-Waller factors via -buni later in the MSA program. |